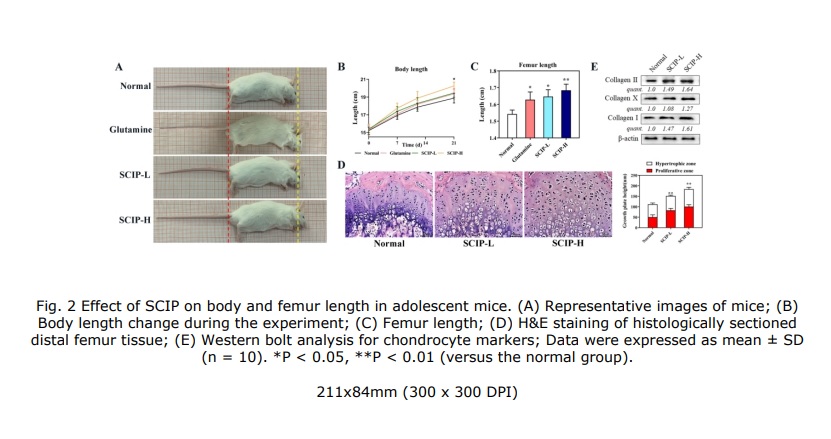
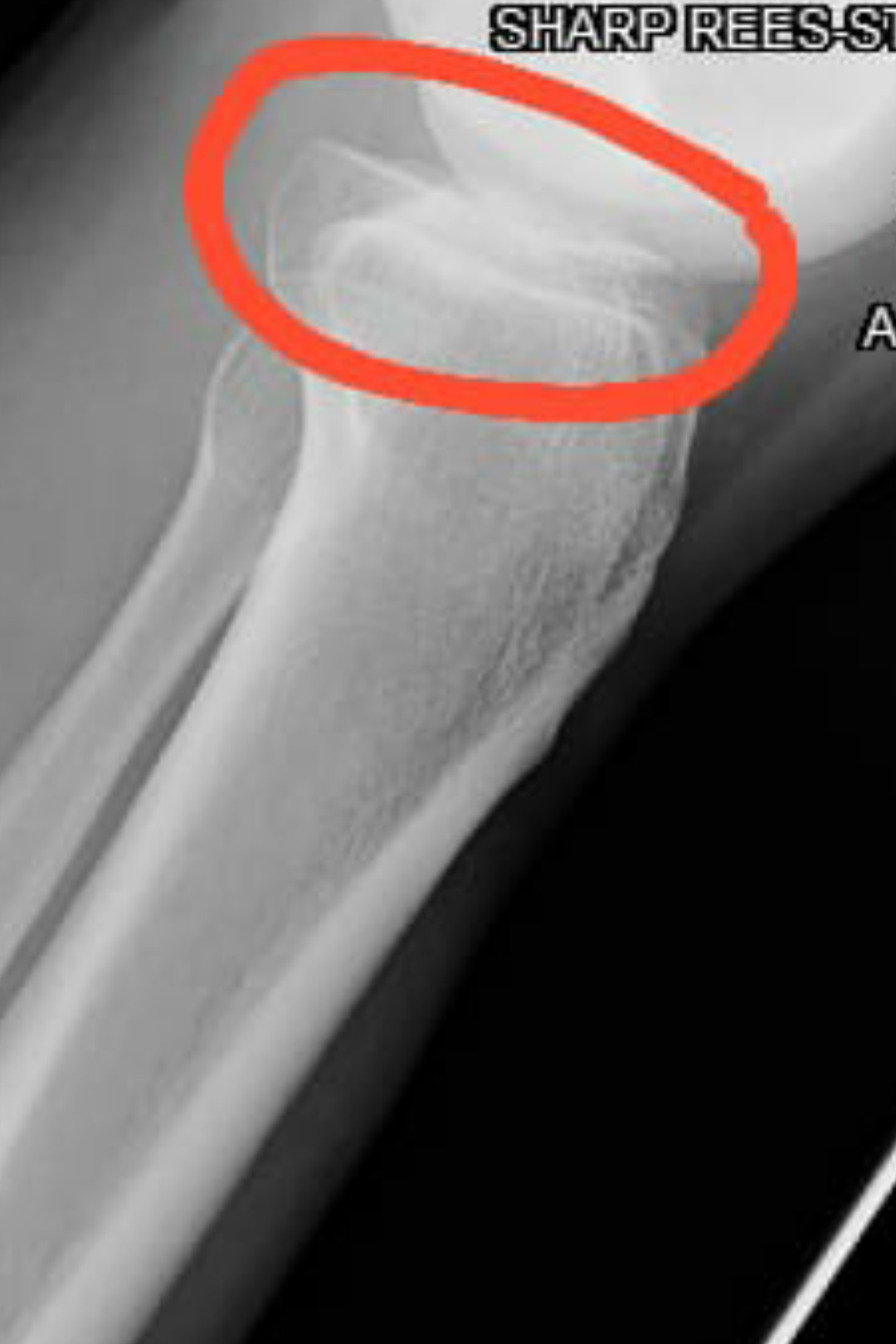
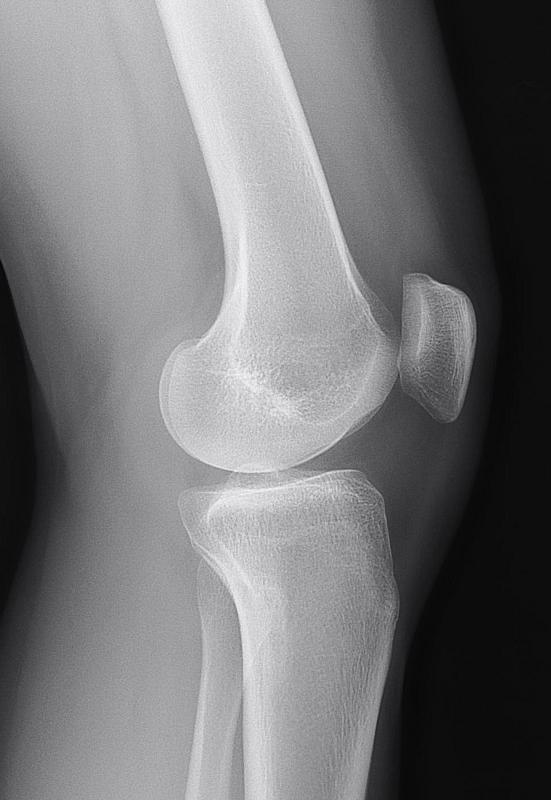
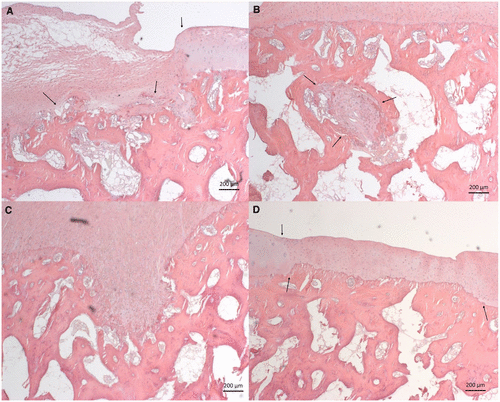
New study related to lateral synovial joint loading:
Knee Loading Enhances the Migration of Adipose‑Derived Stem Cells to the Osteoarthritic Sites Through the SDF‑1/CXCR4 Regulatory Axis
“Osteoarthritis (OA) is a whole joint disorder that is characterized by cartilage damage and abnormal remodeling of subchon�dral bone. Injecting adipose-derived stem cells (ASCs) into the knee joint cavity can assist in repairing osteoarthritic joints, but their ability to migrate to the damaged site is limited. Our previous studies have shown that knee loading can improve the symptoms of OA, but the effect and mechanism of knee loading on the migration of ASCs in OA remain unclear. We employed a mouse model of OA in the knee and applied knee loading (1 N at 5 Hz for 6 min/day for 2 weeks) after the intraarticular injection of ASCs. The cartilage and subchondral bone repair were assessed by histopathological analysis. Immunofuorescence assays were also used to analyze the migration of ASCs. Using cell cultures, we evaluated the migration of ASCs using the transwell migration and wound healing assays. In vivo experiments showed that knee loading promoted the migration of ASCs, increased the local SDF-1 level, and accelerated the repair of the OA-damaged sites. Mechanistically, the observed effects were blocked by the SDF-1/CXCR4[SDF-1/CXCR4 promotes neovascularization which is great] inhibitor. The in vitro results further revealed that knee loading promoted the migration of ASCs and the inhibition of SDF-1/CXCR4 significantly suppressed the beneficial loading effect. The results herein suggested that the migration of ASCs was enhanced by knee loading through the SDF-1/CXCR4 regulatory axis, and mechanical loading promoted the joint-protective effect of ASCs”
Now we don’t know how much more lateral knee loading stimulates neo-vascularization etc. than regular exercise. Nor do we know if knee loading promotes subchondral bone repair independently of adipose derived stem cells. But neovascularization and new cartilage formation are very important findings that could potentially be linked to what’s needed to grow taller.
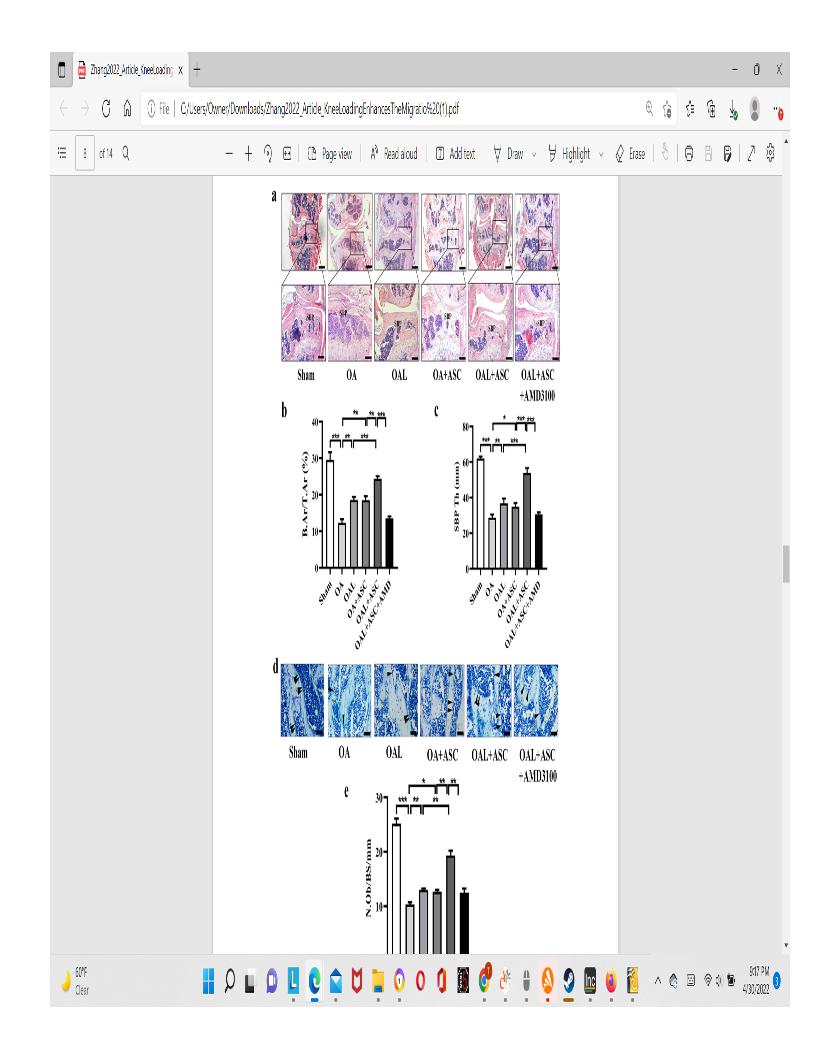
“A variety of physical rehabilitation regimens can afect the remodeling process of articular cartilage and subchon�dral bone and alter the symptoms of OA, such as whole-body vibration, axial loading, and bending loading. The knee loading modality, developed by our laboratory, is a mild non-invasive physical therapy, which applies transverse loading to a synovial joint, such as the elbow, knee, and ankle. Knee loading has been shown as an effective loading option for improving OA symptoms. Previous studies showed that knee loading reduced MMP13 activity and prevented OA-induced cartilage degeneration through cross-talk of the cartilage with subchondral bone. Mechanical loading also mitigated OA symptoms by regulating the stress to the endoplasmic reticulum and autophagy. Further, knee loading facilitated repairing osteoporotic OA by relieving the abnormal remodeling of the subchondral bone via Wnt/β-catenin signaling. It contributed to promoting the diferentiation of MSCs into osteogenic and chondrogenic fates”
“Stromal cell-derived factor 1 (SDF-1), a chemokine of a CXC family, and its receptor CXC chemokine receptor 4 (CXCR4) exist widely in the body, and they regulate a variety of physiological processes, including cell proliferation, migration, adhesion, differentiation, and wound healing ”
“A total of 108 female C57BL/6 mice (14 weeks old, Animal Center of Academy of Military Medical Sciences, China) were used.”<-not quite as old as we would like.
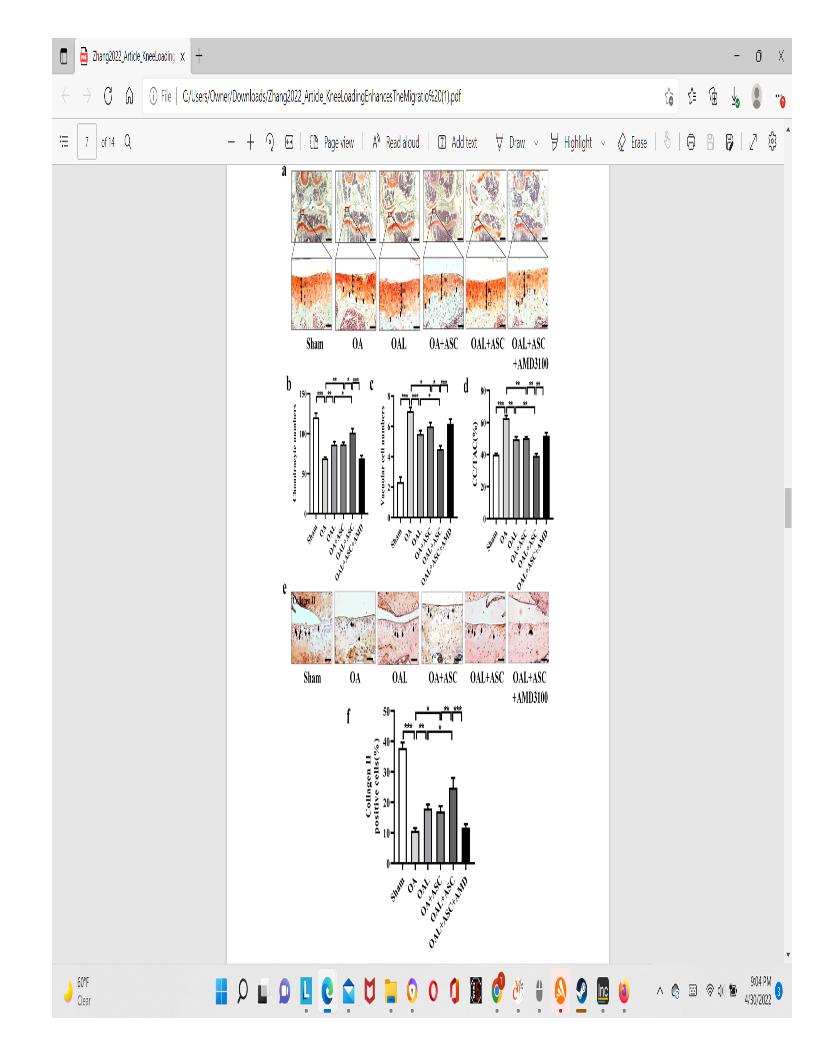
“After the induction of OA, mice were injected with GFP-ASCs, followed by daily knee loading for 2 weeks. The injected ASCs in the knee joints of all groups were identified by immunofuorescence staining with antibodies against GFP. In vivo studies showed that GFP-positive cells were found in the OA cartilage 2 weeks after their injections”
” The number of chondrocytes and vacuolar cells was counted. The number of chondrocytes in the OA group was significantly decreased compared to the sham-treated control group, but the number was restored by knee loading or the injection of ASCs”<-it’s very promising that the chondrocyte number was restored be knee loading independently of stem cell injection but again these mice are not as old as we would like.
” IHC analysis showed that collagen II-positive cells in the OA group were significantly decreased. However, the numbers in the OAL group and OA+ASCs group were increased.”<-again very promising that collagen II positive cells were increasing independently of ASC injection
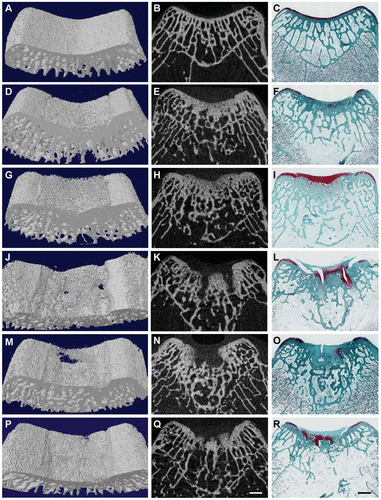
 In the top images you can see that the mice still have their growth plate. It’d be hard to distinguish between changes in the bone caused by Osteoarthritis and those caused by loading. But if you look at the loaded groups(Loading with and without ASC) you can see in red cartilage in places that did not have cartilage in either the control or the osteoarthritis group at the longitudinal ends of the bone. And you don’t really see that in the OA+ASC group. I could be reading it wrong though.
In the top images you can see that the mice still have their growth plate. It’d be hard to distinguish between changes in the bone caused by Osteoarthritis and those caused by loading. But if you look at the loaded groups(Loading with and without ASC) you can see in red cartilage in places that did not have cartilage in either the control or the osteoarthritis group at the longitudinal ends of the bone. And you don’t really see that in the OA+ASC group. I could be reading it wrong though.
 Again here you see cartilage at the sides of the longtiduinal ends of the bone that you don’t really see in the non-loaded groups.
Again here you see cartilage at the sides of the longtiduinal ends of the bone that you don’t really see in the non-loaded groups.