Mice growth plates do not go through the fusion stage but they do cease eventually in longitudinal bone growth due to growth plate dysfunction. If we can reverse growth plate dysfunction in mice than perhaps we can do so in humans and get more height from the growth plate.
This study however does not discuss reversing elderly growth plate dysfunction but rather states that older mice are still capable of undergoing endochondral ossification outside the growth plate.
Fractures in Geriatric Mice Show Decreased Callus Expansion and Bone Volume.
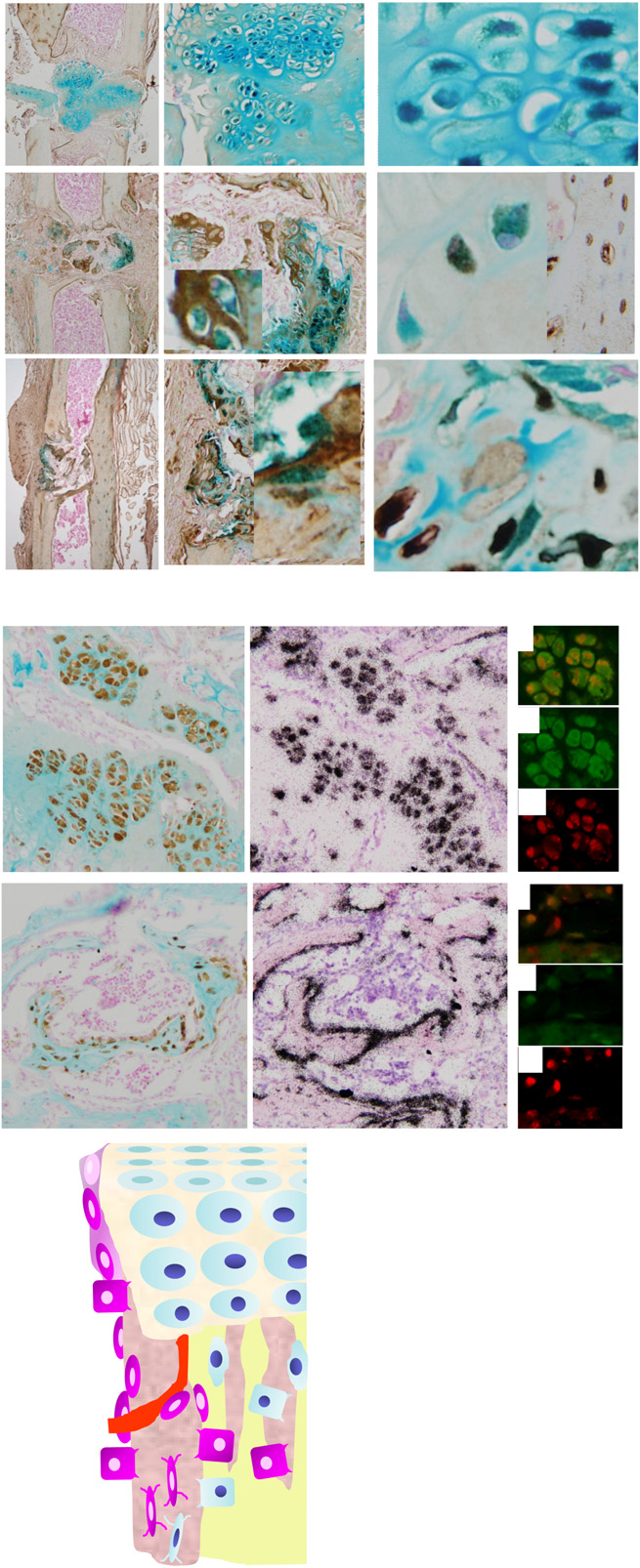
“Using a small animal model of long-bone fracture healing based on chronologic age, we asked how aging affected (1) the amount, density, and proportion of bone formed during healing; (2) the amount of cartilage produced and the progression to bone during healing; (3) the callus structure and timing of the fracture healing; and (4) the behavior of progenitor cells relative to the observed deficiencies of geriatric fracture healing.
Transverse, traumatic tibial diaphyseal fractures were created in 5-month-old (young adult) and 25-month-old (which we defined as geriatric, and are approximately equivalent to 70-85 year-old humans) C57BL/6 mice. Fracture calluses were harvested at seven times from 0 to 40 days postfracture for micro-CT analysis (total volume, bone volume, bone volume fraction, connectivity density, structure model index, trabecular number, trabecular thickness, trabecular spacing, total mineral content, bone mineral content, tissue mineral density, bone mineral density, degree of anisotropy, and polar moment of inertia), histomorphometry (total callus area, cartilage area, percent of cartilage, hypertrophic cartilage area, percent of hypertrophic cartilage area, bone and osteoid area, percent of bone and osteoid area), and gene expression quantification (fold change).
The geriatric mice produced a less robust healing response characterized by a pronounced decrease in callus amount (mean total volume at 20 days postfracture, 30.08 ± 11.53 mm3 versus 43.19 ± 18.39 mm3; p = 0.009), density (mean bone mineral density at 20 days postfracture, 171.14 ± 64.20 mg hydroxyapatite [HA]/cm3 versus 210.79 ± 37.60 mg HA/cm3; p = 0.016), and less total cartilage (mean cartilage area at 10 days postfracture, 101,279 ± 46,755 square pixels versus 302,167 ± 137,806 square pixels; p = 0.013) and bone content (mean bone volume at 20 days postfracture, 11.68 ± 3.18 mm3 versus 22.34 ± 10.59 mm3) compared with the young adult mice. However, the amount of cartilage and bone relative to the total callus size was similar between the adult and geriatric mice (mean bone volume fraction at 25 days postfracture, 0.48 ± 0.10 versus 0.50 ± 0.13), and the relative expression of chondrogenic (mean fold change in SOX9 at 10 days postfracture, 135 + 25 versus 90 ± 52) and osteogenic genes (mean fold change in osterix at 20 days postfracture, 22.2 ± 5.3 versus 18.7 ± 5.2; p = 0.324) was similar{so the deficiencies of older mice to undergo endochondral ossification in response to fracture may be related to things other than gene expression}. Analysis of mesenchymal cell proliferation in the geriatric mice relative to adult mice showed a decrease in proliferation (mean percent of undifferentiated mesenchymal cells staining proliferating cell nuclear antigen [PCNA] positive at 10 days postfracture, 25% ± 6.8% versus 42% ± 14.5%.
the molecular program of fracture healing is intact in geriatric mice, as it is in geriatric humans, but callus expansion is reduced in magnitude.
Our study showed altered healing capacity in a relevant animal model of geriatric fracture healing. The understanding that callus expansion and bone volume are decreased with aging can help guide the development of targeted therapeutics for these difficult to heal fractures.”
So older mice are still capable of chondrogenic fracture healing which is a lot like endochondral ossification. Therefore, this provides evidence that older humans may be capable of inducing new endochondral ossification.
“At 10 days postfracture, corresponding to peak cartilage, young adult mice produced more total cartilage than geriatric mice (302,167 ± 137,806 versus 101,279 ± 46,755 square pixels”
“No difference was found in the percent cartilage content of the callus at 10 days postfracture (41.17% ± 10.13% versus 28.94% ± 8.74%) and in the percent of total cartilage that was hypertrophic at 15 days postfracture (88.76% ± 14.56% versus 70.21 ± 16.05%; p = 0.078). Osseous tissue formation did not reach high levels until 20 days postfracture with cartilage resorption nearly complete for both groups. Geriatric mice exhibit decreased total cartilage production and delayed resorption”