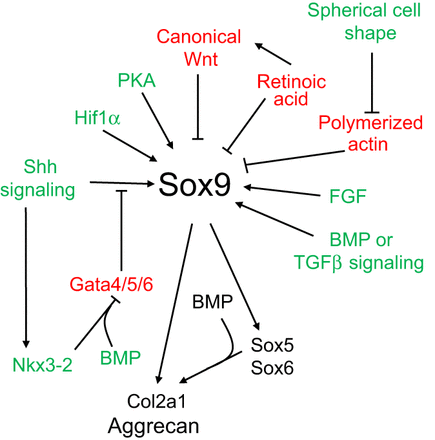
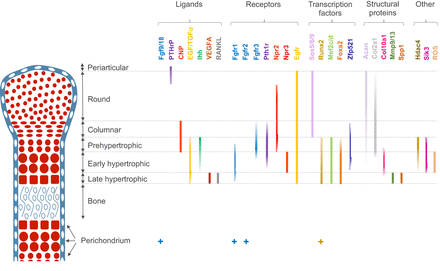
A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation.
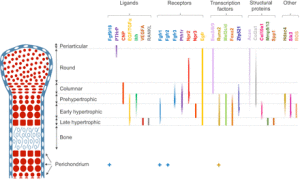
“Mesenchymal progenitors that originate from the cranial neural crest, somites and lateral plate mesoderm contribute to the craniofacial, axial and limb skeleton, respectively. Condensations of such mesenchymal cells express the transcription factor Sox9, which is a key regulator of chondrogenesis, and give rise to cartilage primordia consisting of round immature chondrocytes that continue to express Sox9. Cells lying within the central regions of the cartilage primordia then undergo maturation. During this process, chondrocytes withdraw from the cell cycle and increase ∼20-fold in volume, giving rise to cells that are termed hypertrophic chondrocytes. As the cartilage continues to grow longitudinally, it continually deposits hypertrophic chondrocytes in its wake. These are subsequently replaced by bone in a region known as the primary ossification center. The production and maturation of chondrocytes is eventually restricted to the end of the bone (the epiphysis) in a structure termed the growth plate. A secondary ossification center then arises within the epiphysis, separating the growth plate from the distal ends of the long bones. ”
“Within the growth plate, small round distally located chondrocytes initially give rise to flattened chondrocytes that are more centrally located in the cartilage primordia{You can see the flattened and round chondrocytes in the LSJL loaded growth plates}, and which proliferate and stack into longitudinal columns. These immature chondrocytes express the transcription factors Sox5, Sox6 and Sox9, and the structural proteins collagen, type II, α1 (Col2a1) and aggrecan (Acan). The next stage of maturation into prehypertrophic chondrocytes is marked by the expression of both parathyroid hormone 1 receptor (Pth1r) and Indian hedgehog (Ihh). This is followed by maturation into early hypertrophic chondrocytes that express collagen, type X, α1 (Col10a1). Notably the induction of Pth1r, Ihh and, subsequently, Col10a1 correlates with the loss of Sox5, Sox6, Sox9, Col2a1 and Acan expression. Finally, Col10a1-expressing cells lose expression of this collagen and progress to become late hypertrophic chondrocytes, which express vascular endothelial growth factor A (VEGFA), matrix metalloproteinase 13 (Mmp13) and secreted phosphoprotein 1 (also known as, osteopontin/bone sialoprotein 1; Spp1). VEGFA and Mmp13 expression herald the invasion of the growth plate by endothelial cells, osteoclasts and osteoblast precursors. Osteblast precursors that arise from both the perichondrium and the hypertrophic chondrocytes work together with osteoclasts to remodel the growth plate matrix to form trabecular bone. ”
“Limb bud mesenchymal cells can undergo chondrogenesis in culture when plated at extremely high density (termed micromass culture) or at low density but only when these cells are plated in either suspension culture or collagen gels. This leads to speculation that a spherical cell shape might promote chondrogenesis”
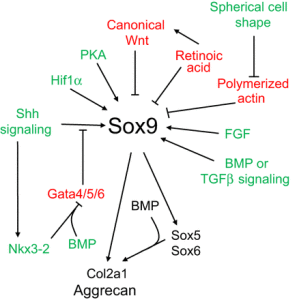
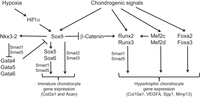
“During development, the somite undergoes a process of differentiation. The ventral region becomes mesenchymal and forms the sclerotome, precursor to the vertebrae and to the medial part of the ribs. A signaling molecule known to be crucial for sclerotome induction is sonic hedgehog (Shh), a member of the hedgehog family of proteins that is expressed by both the notochord and the floorplate of the neural tube. Interactions between Shh and the patched transmembrane receptors Ptch1 and Ptch2 cause another transmembrane protein, smoothened (Smo), to translocate to the base of the primary cilium. This leads to activation of Gli transcription factors (Gli1, Gli2 and Gli3), which in turn regulate the expression of hedgehog-responsive genes. Shh signaling has been found to play multiple roles in sclerotome formation: promoting the survival of somitic cells, an epithelial-mesenchymal transition of the sclerotome via induction of Snail and, finally, the induction of sclerotome-specific markers such as Pax1{LSJL upregulates Pax1}, Sox9 and Nkx3-2. In addition to Shh, noggin (Nog) (which is expressed in the notochord) and gremlin 1 (Grem1) (which is expressed in the dorsal neural tube and somites) cooperate to maintain a BMP signaling-free zone that is crucial for Shh-mediated sclerotome induction. Mice engineered to lack Shh fail to form vertebrae, indicating that Shh signaling is crucial for the formation of vertebral cartilage. the subsequent differentiation of sclerotome into cartilage does not depend on maintained Shh signaling. Shh signals induce the expression of Sox9 and the transcription factor Nkx3-2, which indirectly maintains Sox9 expression in somitic cells. Bone morphogenetic protein (BMP) signals maintain the expression of these genes after their initial induction by Shh, although they cannot induce the expression of either Sox9 or Nkx3-2 in presomitic paraxial mesodermal cells that have not yet been exposed to Shh signaling. Provided that BMP signals are present, Sox9 can both regulate its own expression and induce expression of Nkx3-2. Nkx3-2 is a BMP-dependent transcriptional repressor that blocks the expression of inhibitor(s) of Sox9 transcription. Nkx3-2 blocks BMP-dependent expression of several GATA transcription factors (Gata4, Gata5 and Gata6) in explants of paraxial mesoderm, and these GATA factors can in turn block Shh-dependent induction of Sox9 gene expression ”
“conditional loss of β-catenin expression in either limb or head mesenchymal progenitors both increases the expression of Sox9 in these progenitor cells and induces chondrocyte formation at the expense of osteoblasts“<-So a Beta-Catenin inhibitor may be a useful height increase tool.
“In addition to Wnts secreted by the limb bud ectoderm, FGFs secreted by the apical ectodermal ridge (AER) are necessary to maintain: (1) limb bud outgrowth; (2) the viability of a chondrogenic precursor pool that gives rise to the cartilage templates of the limb; and (3) the competence for limb bud mesenchymal cells to undergo chondrogenesis once the Wnt signals are removed. In addition, FGF signals have been demonstrated to boost the expression of Sox9 in primary chondrocytes via a mitogen-associated protein kinase (MAPK)-dependent pathway ”
“Wnt signals induce both a repressive chromatin mark (H3K27me3) and DNA methylation over the Sox9 promoter, and that Wnt-induced irreversible silencing of the Sox9 gene requires DNA methylation of this locus that is specifically countered by FGF signals. FGF blocks the recruitment of the de novo DNA methyltransferase DNMT3A to the Sox9 promoter by inducing the interaction and phosphorylation of DNMT3A by extracellular-regulated kinase (ERK) 1 and ERK2, and thereby controls whether the expression of Sox9 is irreversibly or reversibly silenced by Wnt signals in limb bud mesenchymal cells{This step may be the reason that increased FGFR3 signaling reduces height}”
“Wnts secreted by the ectoderm act via a β-catenin-dependent pathway to block Sox9 expression and cartilage formation in limb bud mesenchymal cells, and that FGF signaling maintains chondrogenic competence of these cells by blocking DNA methylation of the Sox9 promoter{So neo-growth plate formation in adult bone MSCs may be partially blocked by DNA methylation of Sox9 in these cells but MSCs are definitely capable of chondrogenic differentiation as shown by distraction osteogenesis}.”
Blocking TGF-Beta signaling did not appear to block the initial stages of chondrogenesis.
“GFβ signaling inhibition has limited effects on the formation of Sox9+Scx− progenitors, TGFβ signaling is absolutely necessary for both formation of the Sox9+Scx+ progenitor population and the subsequent development of bone eminences”
” The mis-expression of BMPs or of activated BMP receptors in the limb bud results in ectopic chondrogenesis”
“Hif1α was shown to promote the expression of Sox9 and to elevate the expression of glycolytic enzymes and glucose transporters to allow chondrocyte differentiation and adaptation to hypoxic conditions. Hif1α was also shown to upregulate VEGFA expression in the growth plate as well as promote collagen hydroxylation to enable collagen secretion by hypoxic chondrocytes”
“Sox9 loss of function leads to premature maturation of immature chondrocytes into hypertrophic cells. Conversely, overexpression of Sox9 in either immature or hypertrophic chondrocytes of the growth plate slows the process of chondrocyte hypertrophy”
“the ectopic expression of Runx2 in immature chondrocytes drives premature cellular maturation and induces the expression of Col10a1 and other hypertrophic markers”
“Mef2c loss of function in chondrocytes results in shortening of the bones, a delay in chondrocyte hypertrophy and downregulation of Runx2 expression. Conversely, gain-of-function experiments in transgenic mice that express an activated Mef2c-VP16 fusion protein in chondrocytes results in the opposite phenotype of premature and excessive endochondral ossification”

“Ihh promotes chondrocyte proliferation, at least in part by increasing the expression of cyclin D1, which regulates cell cycle progression. In addition, Ihh promotes the transition of small round chondrocytes into proliferating chondrocytes, independently of PTHrP expression (Kobayashi et al., 2005), by inhibiting the repressor activity of Gli3 (i.e. Gli3R)”
“The mechanical regulation of chondrocyte proliferation affects not only the length of the bone but also its morphology. For example, both the ‘mini’ growth plate of bone eminences and the endochondral ossification-mediated repair of bone fractures are controlled by muscle loading”